
By Kelly Patricia O’Meara
January 7, 2016
The Food and Drug Administration (FDA) has initiated a proposal that would reclassify Electroconvulsive Therapy Devices (ECT) from its highest risk category III to allow electric shock machines to be utilized in the treatment of specific alleged mental illnesses with less regulatory controls.[1] This is despite the federal agency’s admission that the ECT device has not been proven safe and effective. To date, nearly five million Americans have received ECT “treatment” without ECT manufacturers being required to submit valid scientific evidence, such as clinical trials, of the device’s safety or effectiveness.[2]
The proposal has reignited a firestorm that the FDA has colluded with the American Psychiatric Association (APA) to promote a dangerous treatment and protect the fiscal concerns of APA members rather than protect patient lives.
It is no secret that the FDA has procrastinated for more than five years over this proposal after first entertaining the idea of reducing the device’s risk category in 2009, when it requested public input on this. At a public hearing in January 2011, it was heard that nearly 80 percent of the respondents and a further 92 group submissions representing more than 6000 individuals, were against reclassification.[3]
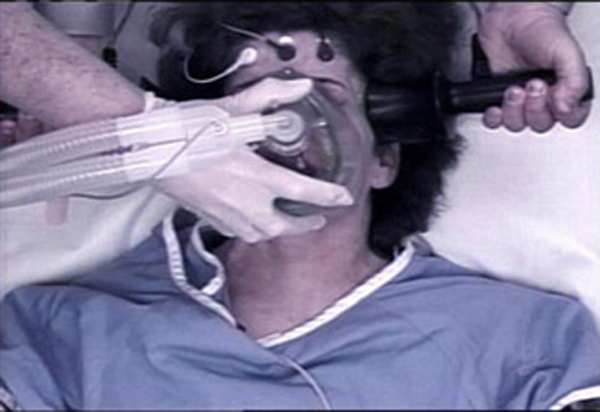
ECT is the practice of up to 460 volts of electricity sent searing through the brain, with no scientific evidence to substantiate how this procedure “works.” Evidence and patient reports reveal that ECT causes memory loss, cardiovascular complications, brain damage and death. In 1979, the FDA classified the ECT device as Class III because of its “potential unreasonable risk of illness or injury.”

ECT is the practice of 460 volts of electricity sent searing through the brain, with no scientific evidence to substantiate how this procedure “works.”
The manufacturers have never been required to provide a Premarket Approval application (PMA) with clinical trials to prove the device is safe. In 1990 and 2009, the Federal government required the FDA to either reclassify Class III medical devices or ensure manufacturers submitted PMAs if they remained in the high risk classification. ECT device manufacturers have failed to submit any PMAs.
Despite seriously outrageous risks associated with ECT, the FDA’s current proposal would reduce the risk classification of ECT devices for treating “severe major depressive episode (MDE) associated with major depressive disorder (MDD) or bipolar disorder (BPD) in patients 18 years of age or older who are treatment-resistant or who require a rapid response due to the severity of their psychiatric or medical condition.”
While having no clinical evidence to support the safety or efficacy of the device, the FDA assumes that “special controls…together with general controls applicable to the devices would provide reasonable assurance of safety and effectiveness.” The FDA further “believes that…the probable benefit to health from the use of the device outweighs the probable injury or illness from such use.” The FDA “acknowledges significant risks associated with ECT, but believes that for the specified population…the probable benefit of ECT outweighs these risks.” [Emphasis added]
Reasonable assurance? Probable benefit? Sounds very like psychiatry’s unproven, but lucrative depression “may be due to a chemical imbalance” theory, used to reap profits in the tens of millions of dollars in antidepressant sales, just as psychiatry already has profited from ECT’s $1.2 billion a year industry.
Organizations such as the Citizens Commission on Human Rights (CCHR) argue that the FDA’s “split classification” (Class III for some disorders and Class II for MDE, MDD and BPD) is illogical—that there is no evidence that these specified disorders exist in the brain whereby electricity can selectively target and not damage, as opposed to applying it to other “disorders” that it can damage. “If two people with different mental disorders were to stick their fingers in an electrical socket, wouldn’t the damaging outcome be the same?” asked CCHR’s president, Jan Eastgate. “There’s no justification for what the FDA is proposing and, if successful, there are no controls over how psychiatrists will abuse this. Tens of thousands could be further harmed every year.”

The health risks associated with ECT include the following: Death may result from various complications of ECT; there can be cardiovascular complications; cognition and memory impairment; the device can malfunction which may cause electrical hazards, such as prolonged seizures and skin burns; physical trauma; prolonged or tardive [latent] seizures; and pulmonary complications.
The FDA lists fourteen health risks associated with ECT, including the following:
Death may result from various complications of ECT such as reactions to anesthesia, cardiovascular complications, pulmonary complications, or stroke (impairment of blood flow to the brain or bleeding in the brain).
Cardiovascular complications. The induced convulsions may be accompanied by arrhythmias (irregular heartbeat) or ischemia/infarction (i.e., heart attack). Hypertension (high blood pressure) and hypotension (low blood pressure) may occur.
Cognition and memory impairment, specifically immediate post-treatment disorientation, anterograde memory impairment and retrograde personal (autobiographical) memory impairment.
Device malfunction: Faulty hardware, software or accessories (electrodes) or improper use may cause electrical hazards, such as the risk of excessive dose administration, prolonged seizures, and skin burns.
Physical trauma: Injury from unconscious violent movements during convulsions (i.e., when drugs administered with the procedure to quell the convulsions fail).
Manic symptoms: ECT treatment may result in the development of hypomanic or manic symptoms.
Prolonged or tardive (delayed) seizures may ensue, and status epilepticus (continuous unremittent seizures) may occur if prolonged seizures are not properly treated.
Pulmonary complications may result, such as prolonged apnea (no breathing) or inhalation of foreign material, such as regurgitated stomach contents.
Worsening of “psychiatric” symptoms.
One has to wonder, then, why has such a possibly deadly device been allowed for decades to be utilized and never undergone clinical trials for safety and efficacy? Further, from what source will the patient obtain a “detailed summary of clinical testing pertinent to the use of the device…”? Clinical testing has never occurred on the devices.
More confounding is the point made in the FDA proposal that “absent the special controls identified in this proposed order, general controls applicable to the devices are insufficient to provide reasonable assurance of the safety and effectiveness of the device.”
In essence, the FDA believes ECT machines now will be safe and effective simply by virtue of several “special controls.” The “controls” only include labeling provisions (additional disclosures and instructions being provided to physicians and would-be patients), establishing a few “technical parameters,” and implementing a few points to ensure devices don’t malfunction.
At the same time, the FDA is admitting that it has allowed millions of Americans to be subjected to electroshock without “special controls,” thereby willfully putting people at risk—a fact that warrants Congressional investigation into both the FDA’s failure to protect the people it serves and any conflicts of interest the federal drug agency has with the ECT device manufacturers or APA that may have encouraged this abuse.

CCHR is a non-profit, public benefit organization.
Kelly Patricia O’Meara is an award-winning former investigative reporter for the Washington Times’ Insight Magazine, penning dozens of articles exposing the fraud of psychiatric diagnosis and the dangers of the psychiatric drugs—including her ground-breaking 1999 cover story, “Guns & Doses,” exposing the link between psychiatric drugs and acts of senseless violence. She is also the author of the highly acclaimed book, Psyched Out: How Psychiatry Sells Mental Illness and Pushes Pills that Kill. Prior to working as an investigative journalist, O’Meara spent sixteen years on Capitol Hill as a congressional staffer to four Members of Congress. She holds a B.S. in Political Science from the University of Maryland.
References:
[1] http://www.fda.gov/AboutFDA/Transparency/Basics/ucm194438.htm.
[2] Department of Health and Human Services, Food & Drug Administration, “Neurological Devices; Reclassification of Electroconvulsive Therapy Devices Intended for Use in Treating Severe Major Depressive Episode in Patients 18 Years of Age and Older Who Are Treatment Resistant or Require a Rapid Response; Effective Date of Requirement for Premarket Approval for Electroconvulsive Therapy for Certain Specified Intended Uses,” Federal Register, Vol. 80, No. 249, 29 Dec 2015, Proposed Rules, 21 CFR Part 882 [Docket No. FDA–2014–N–1210], https://www.gpo.gov/fdsys/pkg/FR-2015-12-29/pdf/2015-32592.pdf.
[3] FDA Executive Summary, Prepared for the January 27-28, 2011 meeting of the Neurological Devices Panel, Meeting to Discuss the Classification of Electroconvulsive Therapy Devices (ECT), http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/MedicalDevices/
MedicalDevicesAdvisoryCommittee/NeurologicalDevicesPanel/UCM240933.pdf.